

峰瑞报告第 15 期,我们来聊聊成功率仅 3% 的药物研发。
二战中,青霉素的发现与使用,拯救了不计其数的伤员。它帮助人体抵抗细菌感染,成为士兵口中的 “救命药”、丘吉尔眼中 “二战时最伟大的发明”。
电影《我不是药神》中的神药格列卫,超过 96% 的新发慢性骨髓性白血病患者服用后完全响应,五年疾病无进展生存率从 64% 提高到 83%,几乎与正常人无异。
诸如此类的药物研发突破和进展,让人类的疾病得以治愈、生活质量提升、寿命延长。不过,已有 100 多年的药物研发史上,还有包括老年痴呆症在内的许多难题未被攻克;悲剧也从未缺席,比如,沙利度胺的胎儿致畸作用导致了 “海豹儿” 事件。
做药就像玩高空抛环一样,需要在活性、选择性、可成药性、代谢、安全性之间找到平衡。在找到平衡的路上,不计其数的临床前研究和临床试验折戟沉沙,几百亿美金打了水漂也是常态。
这篇报告将从科普的角度,和你一起回顾药物研发史上的关键事件和节点,并为你解读药物研发的历程。你将了解:

-
为什么说药物研发是长周期、高投入、高风险的冒险?
-
为什么开发一款药那么难,为什么很多药定到天价?
-
小鼠、大鼠、狗和猴子,还有兔子、猪、土拨鼠、鸭子……这些模式动物为药物研发做了什么贡献?
-
当我们吃下一粒药片,我们的身体对药物做了什么,药物又对我们的身体做了什么?
如果说在药物研发领域创业,烧钱又冒险,那么,在这个领域做早期投资,同样意味着昂贵、漫长、充满风险和不确定性。不过,除去商业上的考量,支持医药创新是一项有社会意义的事业。峰瑞长期看好对包括新药研发、医疗器械、体外诊断和医疗服务在内的医疗健康领域的投资,并且有足够的耐心和资源来陪伴初创公司成长。期待与更多医疗领域的创业者交流、探讨。

药物研发历程
文 / 王一恺 (yikai@freesvc.com)
/ 01 /
生命不息,制药不止
在 2017 年、2018 年两年全球销售额 Top10 的创新药中,有 8 款生物药(7 个抗体药和 1 个融合蛋白)和 2 款化学药。
来认识下这些创新药中销售额超过 10 亿美金的 “重磅炸弹” 们:药王修美乐(Humira,阿达木单抗)蝉联榜首,2017 年销售额达到 184 亿美金。这是一款生物药,由艾伯维公司研制。第二名是化学药瑞复美(Revlimid,来那度胺),销售额达到 81 亿美金,由新基公司研制。第三名是恩利(Enbrel),由安进公司研制……
遗憾的是,还没有一款 “重磅炸弹” 出自中国公司。
峰瑞观点(freesvc)
- 和个人及家庭相关的医疗消费近年来不断增加。2015 年,中国居民医疗消费总额 4 万亿,并以每年约 20% 的速度增长。重大疾病的诊断和用药,占了居民医疗消费的大头,其中最大的一块是进口药品和进口医疗器械。要解决可及性和负担得起的问题,除了平抑流通过程中不合理的价格,还要想尽办法实现关键技术和产品的国产化。
虽然生物药的研发和销售是当下的主流,现代药物研发的历史却是从化学药开始的。
按照药物的成分和组成,大体可以分为:
化学药:起效成分单一、明确的小分子药物,分子量通常小于 1000 道尔顿
生物药:抗体、蛋白/多肽、核酸类药物,分子量通常远大于 1000 道尔顿
生物制品:成分非单一的疫苗、病毒和细胞类药物
按照药物的研发和商业化阶段,大体可以分为创新药和仿制药:
创新药:是指具有自主知识产权的药物
仿制药:指对创新药的仿制品,一般在创新药专利保护期到期后,仿制药才可以允许上市
/ 02 /
从 19 世纪中叶至今
药物研发途径的演化
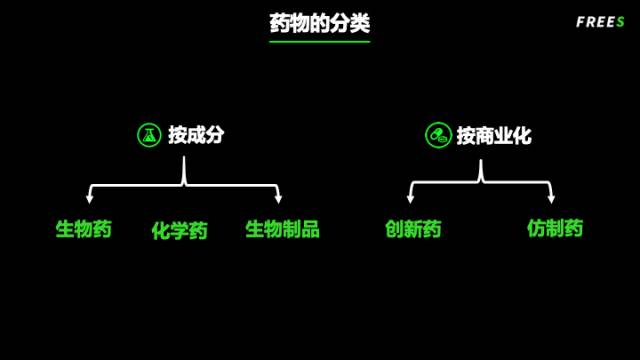
我们以小分子化学药为例,来回顾一下药物研发的三个阶段。
第一个阶段是基于化合物的时代(compound based),大概从 19 世纪中叶制药工业兴起到上个世纪 50、60 年代分子生物学普及之前,都没有靶点这个概念,有的只是从自然提取或人工合成的化合物。从这些化合物中,找到有生物活性的那些,再去研究能治什么病。(详见盘尼西林奇迹、沙利度胺的悲剧与逆袭)
第二个阶段是基于靶点的时代(target based)。这个时代的开端是上个世纪 70、80 年代,与分子生物学、高通量筛选和组合化学一同兴起。这种模式的起点是选出一个生物机理相对明确的 “靶点”,筛选得到可以调节该靶点的化合物,进行成药性优化,最终在临床试验中加以验证。(见“神药” 格列卫、阿尔茨海默症赌局)
第三个阶段是基于病人的时代(patient based)。这一阶段的大门随着 2000 年人类基因组测序完成而打开。核心是从病人出发找到缺陷,然后针对缺陷进行药物研发,可以依赖靶点,也可以不依赖具体靶点,最终治疗疾病。大家平常听到的个体化医疗、精准医疗、基因治疗等词汇,都是这个时代的关键词。不过,这个阶段才刚刚开始,前面的路还很长。(见吉非替尼——上帝给东方人的礼物、“篮子” 试验的成功)
/ 03 /
药物研发史上的奇迹与遗憾
每一个阶段,都有突破和曲折,悲喜剧始终在上演。
▍盘尼西林奇迹
盘尼西林,即青霉素的发现,是第一阶段基于化合物时代的经典案例。

▲ 二战士兵们的 “救命药”。
1928 年,英国细菌学家弗莱明发现了一种新霉菌,其周围的葡萄球菌菌落被溶解无法生长。当时同事们认为这只受到污染的培养皿应该废弃,但弗莱明认为值得研究。最终,他确认这是一种尚未被人类发现的物质,有着极强的杀菌作用。因为是青霉分泌,就被命名为青霉素。
不过,弗莱明没能成功提纯出这种物质。直到 1941 年,弗洛里与钱恩实现了对青霉素的分离与纯化,1942 年实现了大规模生产,青霉素于 1943 年获批上市。
在第二次世界大战中,正是由于青霉素的发现与使用,无数人被拯救。也是因为抗生素领域的研究进展,人类的预期寿命大大延长。
▍“神药” 格列卫
在基于靶点的药物研发时代,一个最近被大众熟知的药物,就是出现在电影《我不是药神》中的格列卫(格列宁的原型)。其被用于治疗慢性髓系白血病,2001 年获批上市。
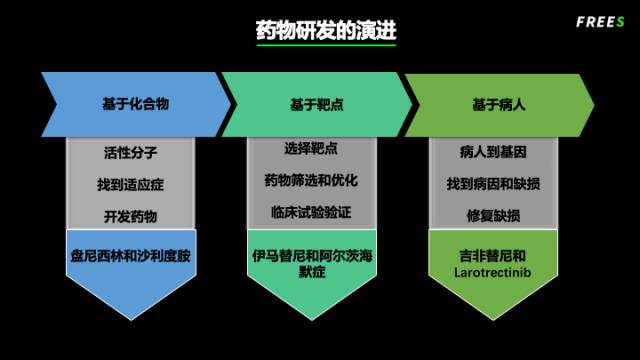
格列卫被称为药物史上的奇迹之一。1960 年代,宾夕法尼亚大学病理系教授彼得·诺维尔(Peter Nowell)发现,慢性髓系白血病患者的癌细胞染色体中,第 22 号染色体明显要更短。1973 年,芝加哥大学的珍妮特·罗利(Janet Rowley)教授发现,第 22 号染色体之所以短,是因为发生了染色体的易位——9 号染色体与 22 号染色体发生了一部分的交换。
1987 年,科学家们进一步发现,由于染色体的交换产生了一个新的蛋白叫 BCR-ABL,其在正常细胞中原本并不存在。这个新蛋白的出现,导致细胞分裂不受控制,引发癌症。
幸运的是,从上世纪 80 年代开始,人们经过一系列的设计与修饰,发现了可以抑制 BCR-ABL 的化合物,它能杀死携带这个突变的细胞。2001 年,这款药物上市,取名伊马替尼,商品名格列卫。
患者的生存曲线证明,这款药物配得上 “奇迹” 这个美誉。超过 96% 的新发慢性骨髓性白血病患者服药后,癌细胞完全消失。相比传统疗法,格列卫将患者的五年疾病无进展生存率从 64% 提高到 83%,几乎与正常人无异。
▍β淀粉样蛋白:阿尔茨海默症赌局
在基于靶点的药物研发登上舞台中心(成为主流)的过程中,人们既收获了礼物,也遭遇了挫折。
至今,人类都没有攻克阿尔茨海默症(俗称 “老年痴呆症”),虽然我们的脚步从未停歇。1984 年,科学家在患有阿尔茨海默症的患者大脑中发现许多 β淀粉样蛋白斑和神经纤维缠结。随后,主流学界认定,聚集在大脑内部的 β淀粉样蛋白是导致阿尔茨海默症的元凶。
于是,人们纷纷押注只要能抑制 β淀粉样蛋白的产生或者降低它的量就能治疗阿尔茨海默症。包括强生、礼来、默克等公司在内的全球制药巨头,都曾在或依然在 β淀粉样蛋白的相关通路上投入巨资。
遗憾的是,截至目前,所有的研发都没有取得实质性进展,无数的临床试验折戟沉沙,几百亿美金打了水漂。2016 年,礼来宣布试验失败。2017 年的情人节,默克宣布没有取得积极成果。2018 年 1 月,辉瑞宣布放弃相关研究。药物研发史上最昂贵的假说和靶点,还在被证实或证伪的路上……
基于靶点的药物研发,成功的关键是该靶点在病理过程中是因还是果。BCR-ABL 是因,从而成就了格列卫,而对于 β淀粉样蛋白与阿尔茨海默症之间的关系,现在更多声音认为,前者是果。
▍吉非替尼——上帝给东方人的礼物
在基于病人的药物研发时代蹒跚而来的过程中,吉非替尼无疑是一个标杆,也是中国临床肿瘤研究第一次登上世界舞台。
1980 年代,随着分子生物学不断发展,人们发现肺癌患者的 EGFR 信号通路存在激活现象。沿着基于靶点的研发思路,针对该蛋白开发出小分子药物吉非替尼,于 2002 年在日本获批上市,取名易瑞沙。2003 年,易瑞沙在美国获批上市,2005 年美国 FDA 又撤销了批准。撤销的原因是,FDA 认为缺乏证据证明吉非替尼显著延长了患者的生命。当时的数据显示,吉非替尼对生存期的延长平均只有两周。这也让肿瘤靶向治疗的价值饱受诟病和质疑。
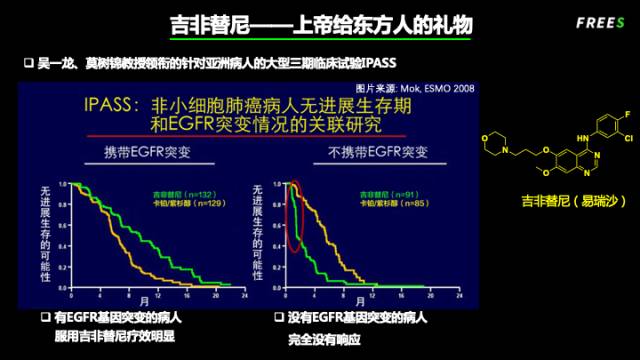
不过,中国学者感到不甘心,广州的吴一龙医生和香港的莫树锦医生在回顾性分析中发现,吉非替尼在肺癌病人中治疗效果确实一般,但在亚洲女性无吸烟史患者中的响应率比较高,这部分病人往往携带 EGFR 基因突变。于是,他们在中国和全亚洲做了一个有划时代意义的临床试验 “IPASS”,对病人进行基因筛选,找出那些携带 EGFR 基因突变的患者,看药物的疗效。
这项研究让全世界震惊,与化疗相比,携带 EGFR 突变的病人服用吉非替尼中位生存期从 6-7 个月延长到了 12 个月;而不携带 EGFR 突变的病人,生存期反而缩短。正是由于这项研究,美国 FDA 于 2015 年重新批准吉非替尼上市,用于治疗携带 EGFR 突变的病人。
▍“篮子” 试验的成功
从病人出发找到病因,针对病因进行药物研发,是第三个阶段的发展趋势,也在慢慢变成现实。在 2018 年 ASCO 会议上,LOXO 公司公布了一项举世瞩目的临床研究结果,在精准医疗发展史上写下了浓墨重彩的一笔。
在几乎所有的常见肿瘤中,都存在 Trk蛋白的融合突变,尽管发生的比例不高。把不同肿瘤中跟 Trk 相关的突变合起来算,一共有十几种新的蛋白产生。如果能像伊马替尼抑制 BCR-ABL 一样抑制这些新的蛋白,就可以抑制肿瘤生长。
有了对病因的理解,接下来的问题可以简化为两方面:第一,能不能找到一个化合物可以同时抑制这十几种不同的蛋白;第二,每一种肿瘤中携带这个突变的比例都很少,如何找到足够的病人开展临床试验。
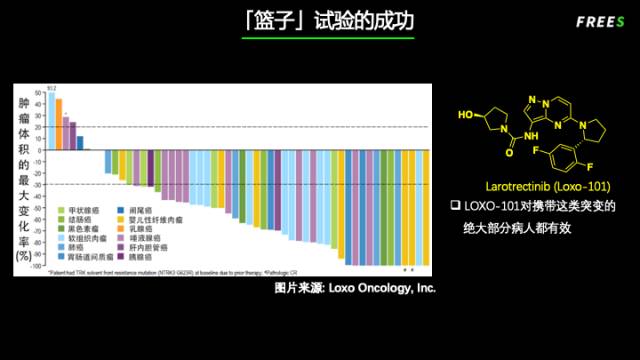
第一个问题,很幸运,这十几种蛋白都是酪氨酸激酶,LOXO 公司找到了仅抑制这些激酶而不抑制其他激酶的小分子 Larotrectinib。
第二个问题,LOXO 公司设计了一种所谓的 “篮子” 试验,就是通过基因测序,把所有肿瘤病人中携带这类突变的病人找出来。不管是胃癌、肺癌、乳腺癌,都放在一起治疗。临床效果非常出色,绝大部分患者的肿瘤都缩小了甚至消失了!LOXO 公司的股票一个月内从 $116 飙到了 $208。
这个 “篮子” 试验之所以至关重要,可以归结为三点,精准性、有效性、经济性。从病人的基因层面出发是为精准,针对的是病理原因所以有效,用 “篮子” 方式增加病人数目可谓经济(保证市场足够大)。
/ 04 /
沙利度胺的悲剧与逆袭
接下来,讲沙利度胺跨越了 60 年的故事。
它被合成出来时,人们还处在基于化合物的药物研发时代,它的风靡一时却造成无数悲剧;它在基于靶点的研发时代问鼎化学药销售额榜首,却一直找不到靶点;当它的靶点被发现后,一个新的方向被开启,为基于病人的药物研发提供了强大的工具。
沙利度胺的胎儿致畸作用导致大量 “海豹儿” 悲剧,可以说是医药史上一段黑暗的日子。

▲ “反应停” 带来的 “海豹儿” 悲剧。
1953 年,诺华的前身 Ciba 合成了一个化合物沙利度胺,本来想用于抗菌,但没有效果。联邦德国的一个药厂发现沙利度胺有一定的镇静催眠作用,还能够显著抑制孕妇的妊娠呕吐。
1957 年 10 月,沙利度胺正式投放到欧洲市场,被包装成 “没有任何副作用的抗妊娠反应药物”,还得名 “反应停”。沙利度胺迅速在欧洲、非洲、澳大利亚和拉丁美洲风靡,大量孕妇使用。
可是,仅仅一年,欧洲的医生们发现,本地区畸形婴儿(海豹儿,上肢发育畸形)的出生率明显上升,并发现这种畸形与沙利度胺息息相关。1961 年,沙利度胺在全世界市场被召回,并于 1963 年正式撤市。截止沙利度胺被召回,全世界约有 15,000 名婴儿受害。德国公司 Chemie Grünenthal 也因此支付了 1.1 亿西德马克的赔偿,被迫倒闭。
就在沙利度胺风靡欧洲时,美国 FDA 拒绝了这一药物的上市申请。FDA 负责这一申请审批的官员弗兰西斯·凯尔西注意到,沙利度胺的人体试验和动物试验数据存在较大差距,她以药理活性不明确,并且人和动物差异大为由拒绝批准。尽管药企向她施压,她还是顶住了压力,避免了悲剧在美国发生。
后来的研究发现,沙利度胺分子是一对镜像异构体,一个异构体有镇静作用,但另一个异构体会导致畸形。在身体中,这两个异构体可以互相转化,所以即使是只服用有镇静作用的那个,到体内也会有一半转化为有致畸作用的那个,这意味着副作用没法避免。
沙利度胺悲剧直接促成了《科夫沃-哈里斯修正案》的通过。主要内容是:确定药品广告申请制度;要求所有药品必须向 FDA 提交安全性和有效性证明;制订新药研究和审批的程序;要求在美药品生产企业必须实施 GMP。
就像硬币的两面,这个法案的通过,在提高药物注册申请上市门槛的同时,也导致新药上市的数量和速度变低。1962 年到 1983 年,没有仿制药上市。此外,因为仿制药也需要做临床试验证明其有效性,这导致药品的价格昂贵、可及性较差。
直到 1984 年,FDA 出台《Hatch-Waxman》法案,提出了仿制药的概念,放宽仿制药门槛,才解决了药物的可及性和成本问题。如今,美国市场大约 80% 的处方是仿制药,但仿制药的销售额只占 30%。
沙利度胺制造了 “海豚儿” 悲剧,只是它命运的开始。沉寂了一段时间之后,“救赎” 的戏码开始上演。
因其免疫抑制活性, 1998 年 7 月 16 日,FDA 批准沙利度胺作为一种治疗麻疯性结节性红斑的药物上市销售。又因其抗血管生成活性,2006 年 FDA 批准沙利度胺与地塞米松连用治疗新发的多发性骨髓瘤患者。后来,Celgene 发现沙利度胺的衍生物来那度胺具有更高的抗肿瘤活性,2008 年,FDA 批准了来那度胺联合地塞米松治疗已经接受过至少一种疗法的多发性骨髓瘤患者。到目前为止,来那度胺在三种肿瘤适应症上获批,进行的临床试验有几十种,年销售 81 亿美金。
故事到这里还没有结束。虽然我们已经进入了基于靶点的药物研发阶段,具有讽刺意味的是,不管是沙利度胺还是来那度胺,它们的作用靶点却一直没有找到。
直到 2011 年,研究人员才发现它们的靶点是 cereblon,一个跟蛋白降解有关的 E3 连接酶。利用这个作用机制,人们又发现可以用小分子选择性地把致病的蛋白降解掉,而实现这种功能目前只能依靠 siRNA 或 CRISPR 技术。从 2014 年开始,小分子蛋白降解剂成为了一个新的热点,有兴趣的读者可以关注一下 PROTAC。这一切的起源,还是那个非常简单的分子——沙利度胺。
从 “海豹儿” 悲剧到在肿瘤领域的应用,再到开启一个新的研发方向,前前后后一共 60 多年时间,故事仍在继续……
峰瑞观点(freesvc)
- 药物研发是一个昂贵、漫长、复杂又充满风险和不确定性的旅程。站在今天回望近两百年的药物研发历史,基础是工具手段的创新和效率的提升,关键是对生命过程的认识和对疾病的理解。当然,这一切最核心的驱动力,是人类对永生的渴望。
/ 05 /
药物研发的九死一生
药物研发是场长周期、高投入、高风险的冒险。有三个 “10” 通常被用来形容这段冒险旅程,耗时 “10年” 成本 “10亿” 美金(近几年上市一个新药,耗时比 10 年长,成本也远超 10 亿美金),当然还有一个 “10” 是用来衡量结果的,即 “10亿” 美金销售额。
为什么会这样?
要理解药物研发的艰难,就要先了解药物研发的流程,这个流程极其庞杂,如下图:
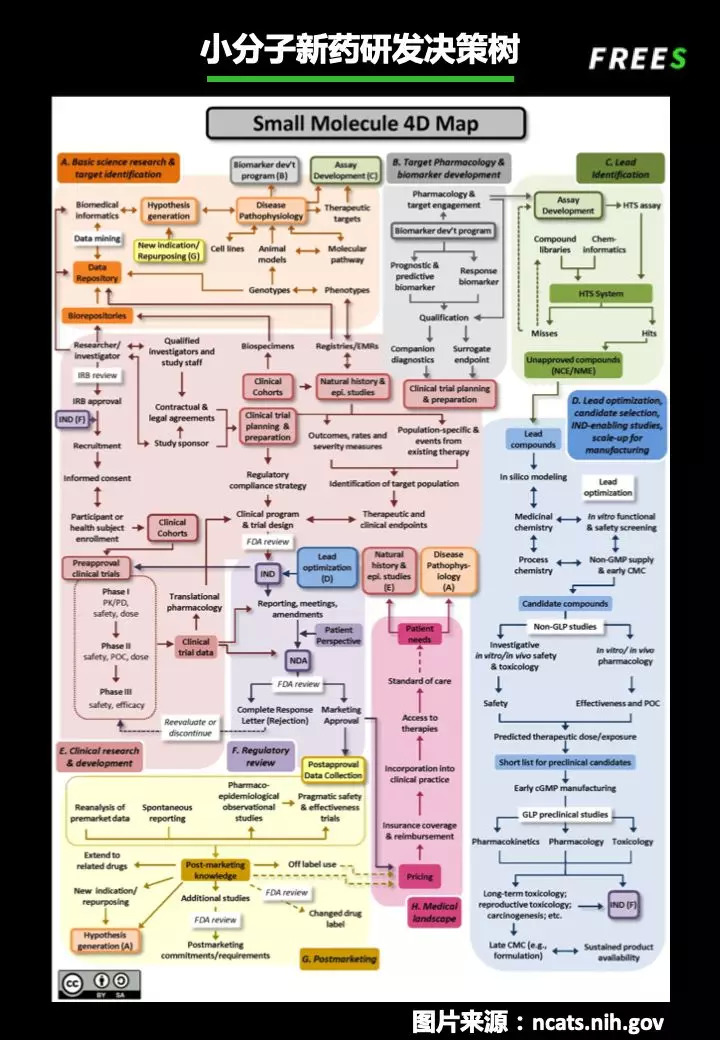
▍从 A 到 H
从早期研发到药物上市,要经历的步骤和过程长到可以从字母 A 排到 H。
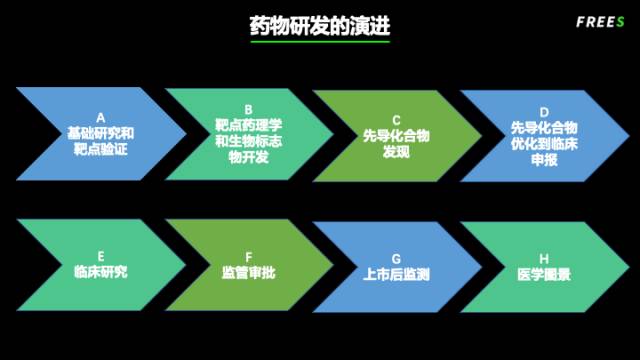
A,首先是基础研究。这个过程就是从病人、临床病理、统计数据入手,找到并验证潜在的靶点。
B,药理学和生物标志物开发。包括靶点的药理学和成药性研究、与靶点和预后相关的生物标志物研究。
C,先导化合物发现。先导化合物是通过各种途径和手段得到的、具有某种生物活性的化合物,通常由体外筛选获得,用于进一步结构改造和优化。
D,先导化合物优化,临床前候选化合物确认,支持申报的临床前研究,生产和质控(CMC)。这个环节中,筛选出潜在的药物候选分子,进行系统的药效学研究和毒理学研究,还需要在符合质量规范管理(GMP)条件下生产出用于支持临床研究的样品。
E,临床研究。主要是进行临床试验,包括一期、二期、三期临床试验。每一期的目标不同,规模也不同。
F,监管审批。这一阶段的主要目标是为药品上市进行注册申报。美国向 FDA 申请,欧洲的审批单位是 EMA,中国则向国家药品监督管理局(CNDA)申请。
G,上市后监测。主要是监测上市后与药物相关的所有信息,包括有效性、安全性、人种差异、非标使用,等等。
H,医学图景。包括竞争情况、定价策略、市场占有率、医生教育等。这些决定了药物的商业成功度。
阿斯利康公司把上述药物研发的流程总结成五个 “R”:正确的靶点、正确的组织、正确的安全性、正确的病人、正确的商业化,并用这五个 “R” 来指导其内部研发和决策,经过 5 年的实践,把项目成功率(完成 III 期临床)从 4% 提升到 19%,远远高于业内平均 3% 的水平。这是非常不容易的事情。

在 A 到 H 的过程中,环节 E 临床试验是投入最大的,也是最容易失败的。为了降低失败率,需要进行大量的临床前研究,为临床阶段降低风险。下面就来说说临床前研究要考虑哪些因素、克服哪些障碍。
▍药物在人体内的漫长旅程
药物经口服以后到发挥作用,通常说有两件事最为重要。第一件事叫药代动力学(Pharmacokinetics),研究的是身体对药物做了什么。第二件事叫药效学(Pharmacodynamics),研究的是药物对身体做了什么。
先来看看药代动力学。下面,让我们跟随口服药一起在身体里做一次旅行。
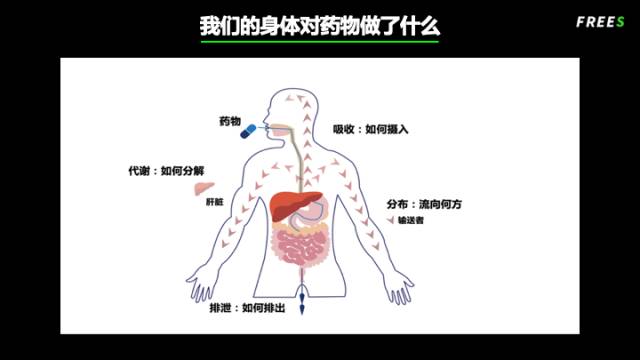
药片口服以后,先在胃部崩解,药片中的化合物就会被释放出来;之后就开始随着胃的蠕动进入到肠。肠是主要的吸收器官,肠壁表面积很大,药物主要在这里被吸收。过肠的时候,肠壁细胞对药物会有代谢,过肠以后经肝门静脉进入肝脏,肝脏是最重要的代谢解毒器官,专门负责清除外源性分子,主要通过肝细胞中的代谢酶对药物分子进行各种各样的修饰,使其失活并被排出。幸存的未被肝脏萃取或修饰的药物分子才最终抵达心脏,经血液输送到全身。这是药物的吸收过程。
药物随血流遍布全身,会扩散进入几乎所有的器官和组织。这个过程称为分布。
药物在不断循环过程中,但凡经过肝脏,都会有一部分被肝脏细胞中的代谢酶修饰。这个过程称为代谢。
最终药物或者修饰过的药物经过胆汁排到肠中随粪便排出,或者经肾脏由尿排出,这是药物的排泄过程。
比如刚才提到的吉非替尼,口服后要足够稳定且被吸收,不会那么快被肝脏代谢,分布到肺中,进入肺癌细胞,接近它的靶点 EGFR,才算最终达到了它的目的地,并开始对人体产生作用,还得保证有足够长的时间和足够高的浓度去发挥作用,也就是我们常说的药物的暴露量。通常药物需要有足够的暴露量才能有治疗效果。
然后,就轮到药效学出场了,它关注的是药物对身体做了什么。药物分子到达靶点附近以后,在多大浓度下能抑制这个靶点的多少活性,抑制了多少活性才能起到药理学作用,需要持续抑制多长时间,如果全天候 24 小时保持完全抑制能达到什么样的疗效,等等。
除了药代动力学和药效学,还要考虑毒性的问题,比如药物分子在正常组织和器官中抑制了这个靶点会不会有副作用,有没有影响到其他不该影响的靶点而带来毒性,药物分子的代谢产物有没有毒性,等等。
身体怎么处置药物是基础;药物对身体产生的影响,正面的是疗效,负面的是毒性;综合起来才能找到一个所谓有治疗窗口的药物,也就是治疗疾病产生的收益大于给身体造成的损害。
▍从模式动物到人
要回答上面说的药物代谢、有效性和毒性等问题,找到一个有治疗窗口的药物,但又不可能把所有的候选分子都在人体中进行测试,就需要进行大量的动物试验。这就要说到药物研发中的模式动物。
常用的模式动物有小鼠、大鼠、狗和猴子,还有不太常用的兔子、猪、土拨鼠、鸭子,等等。
目前,应用模式动物预测药物代谢的能力已经比较高了,也是相对最准确的;在毒性方面,模式动物的预测能力处于中等水平,因为人和动物有显著的差异;在药效的预测上,通常效果都比较差,因为动物模型和人的发病机理、病程都非常不同。
举个例子,美国临床前肿瘤研究年会 AACR 每届都会报道大量好结果,“在小鼠身上又一种什么肿瘤被彻底治愈”,但是临床肿瘤研究年会 ASCO 上,却很少有特别重大的进展。这从侧面反映出肿瘤的小鼠模型到人的转化预测能力比较差。
随着人们对疾病认识的不断加深,以及基因编辑工具的不断迭代,根据病因去构建动物模型,提高转化和预测能力,是目前临床前研究亟待解决的痛点和难点。
▍临床前小分子药物的优化过程
一个药物候选分子进入临床试验之前,需要经过多轮的优化循环。在这个过程中,药物化学家可以改变的是分子结构,即通过体外评价和模式动物的体内评估,来验证结构的改变是不是在某些预期的方面比原来变得更好,以此为基础再进一步做结构修饰和验证……这个循环至少要转几十次,才能找到各方面都可以接受的化合物成为药物候选分子。
在这个优化循环过程中,涉及三方面的关系,结构和活性的关系(与药效有关),结构和性质的关系(与成药性和代谢有关),结构和毒性的关系。
这三个方面的关系,除了做试验测试,还有很多计算和 AI 能帮的上忙的地方。比如,结构和活性的关系,涉及小分子与蛋白的相互作用,如果已知蛋白结构,就可以通过计算机辅助药物设计来进行初步评价,把明显不好的分子淘汰掉。
再比如,结构和性质、毒性的关系,有许多经验和历史数据可以参考。通过大数据和机器学习,可以预测得越来越准确。
这些方向上的工具创新和效率提升,成为最近几年学术界和工业界的研究热点。
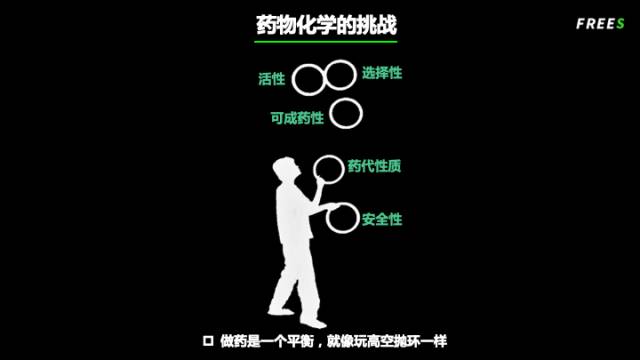
峰瑞观点(freesvc)
- 做药就像玩高空抛环一样,需要在活性、选择性、成药性、代谢、安全性之间找到一个平衡。因为当分子结构改变了,所有性质都会随之改变。这是一个复杂的过程。
/ 写在最后 /
能趟过九死一生并最终成药的幸运分子凤毛麟角,成功率只有 3% 是生命赌局必须面对的残酷现实。这能解释为什么原创新药很贵——一款药的成本,不仅是它自己的成本,还包含了所有失败尝试的成本叠加。许多药定到天价,比如说 100 万美金一个疗程,原因在此。
(欢迎转发至朋友圈。如需转载至其他公众号、网站、移动端 App,请回复 “转载” 了解转载规则,并联系「峰瑞资本」获得授权。版权归峰瑞资本所有。)
评论