

记者 |
编辑 | 许悦
瑞德西韦(Remdesivir)把全国人民对一项临床试验的关注度提升到了史无前例的高度。但随后,被大众给予厚望的瑞德西韦却传出临床试验患者入组不足的消息。
除了瑞德西韦在临床试验上设置的较高门槛外,至今已有接近300项新冠肺炎临床试验在中国临床试验注册中心上登记,这无疑会起到“分流“作用。
但这近300项中,也不乏太极拳、中药注射液、大豆提取物等治疗手段,无论是在医药产业内部还是普通大众中都产生了不少质疑声。
那么,这些“神奇”的临床试验都是如何诞生的,又是如何“一窝蜂”而上乱成了“一锅粥”?
非传统意义的临床试验

实际上,目前开展的绝大多数针对新冠肺炎的治疗性临床试验都并非大众传统认知中的临床试验。
临床试验一般根据发起者不同被分成两种,一种是制药企业申办的研究 ( industry-sponsored trial,IST),另一种则是由研究者或学术机构发起的临床研究( investigator-initiated clinical trial,IIT)。
北京中卫保险经纪总经理曹丽君对界面新闻记者介绍,后者的研究范围往往是前者未涉足的领域,双方并行互为补充,从而更好地推进药物研究的深度和广度,更多地获得研究数据,为循证医学提供依据。
而目前上报至国家药监局药品审评中心(CDE)并获准开展临床试验的药品实际上仅有瑞德西韦、法维拉韦(法匹拉韦)、BDB-001(C5a单抗)与CAStem细胞注射液。
其他无论是氯喹、托珠单抗、克力芝、阿比多尔或者是太极拳等临床试验,都属于“研究者发起的临床研究”。根据申办流程,本就无需上报国家药监局药品审评中心,这是由其自身性质所决定的,并不会单因为审批流程的异同而决定其研究质量高低,如在国外就曾有过许多研究者发起的临床研究取得了重磅研究成果。
之所以会出现研究者发起的临床研究这种形式,药物研发从业者柯楠对界面新闻记者解释了原因——制药企业的逐利性,使得一些已上市的老药品、患者人数少的药品(如罕见病)、或者是患者普遍支付能力差的药品缺乏继续研究的动力。这种情况下,一些医院医生、特定研究机构甚至是监管机构,则会发起相关临床研究。临床研究的范围常常是制药企业申办的研究未涉及的领域,例如罕见病研究、诊断或治疗手段比较、上市药物新用途等,起到填补空白的作用。
但在国内,药品临床试验质量不高早已为业界诟病多年。
2015年后,原国家食药监总局针对制药企业发起的注册性临床试验要求更加严格,但研究者发起的临床试验由于不需要提交药监等监管机构审核,质量依然是参差不齐,难言乐观。
曹丽君认为,研究者发起的研究中,“研究者”既是申办方又是研究者,需要对研究质量、风险管理、受试者权益、财务控制、研究数据的真实性等负责。但是其监管体系模糊。对于研究者发起的项目,并没有明确的管理规定,很多依靠研究者或者机构管理者的经验判断,为管理增加了难度。
在新冠病毒疫情大规模爆发后,短时间内涌现了大量研究者发起的临床研究。缔脉生物副总裁刘熠对界面新闻记者分析,研究者发起的临床研究立项启动相对比较快,在疫情初期能促进快速启动一系列临床试验,探索对抗新冠病毒的、不同严重类型患者的治疗手段,来指导临床诊疗方案的建立。但是,这也很快暴露了突发公共卫生危机压力下临床试验的上马缺乏有效的协调机制的问题。
研究者发起的临床研究立项速度快的原因在于,从流程上看,研究者设计出方案后仅需提交给医院伦理委员会审核通过即可开展研究。
根据2018年修订的《药物临床试验质量管理规范》(GCP),伦理委员会应分别有医药相关专业人员、非科学专业背景人员、非临床试验单位成员,并有不同性别的委员,至少5人组成。所有成员均有伦理审查的培训和经验,能够审查临床试验相关的伦理学和科学等方面的问题。
柯楠表示,为了保护患者安全、尽可能减少风险,伦理委员会实际上承担了类似国家药监局药品审评中心的“把关人”角色,一般来说伦理委员会中还需要有统计师与律师背景人员。
但看着各种一拥而上而又光怪陆离的治疗手段,不得不使人产生怀疑,一些伦理委员会有没有起到“把关人”的作用,或是仅仅是一枚“橡皮图章”。
存在两大问题
刘熠认为,短时间内大量出现的临床试验会造成有限的临床试验资源不能有效分布和利用,比如真正有价值研究的入组速度却得不到保障。由于缺乏有效临床研究管理和足够的临床研究资源,可以预见有部分研究甚至无法按计划完成。
很重要的一个原因是,由于临床试验的发起人一般都仅有临床型专家,对于研究用药本身的药学和临床前数据的专业程度有限,部分药物大跨步进入临床试验阶段,而其立题依据是不够充分的。同时相当一部分的研究样本量很小,研究设计开放性居多,无法提供高质量的临床证据指导用药。
简单而言,药物筛选与临床试验方案设计的不合理,是目前大量的研究者发起的新冠肺炎临床试验所存在的两大问题。
药物筛选主要基于药理来展开,柯楠表示,概念验证(proof-of-concept)与机制证明是在药品拓展适应症时所必须的过程,例如研究瑞德西韦是因为它抗病毒的药物机制,安全性也是得到证明的,研究托珠单抗治疗细胞因子风暴也是因为这款药有获批类似的适应症。
但目前看,许多临床试验都缺乏基本的概念验证,或者其概念验证是备受质疑。
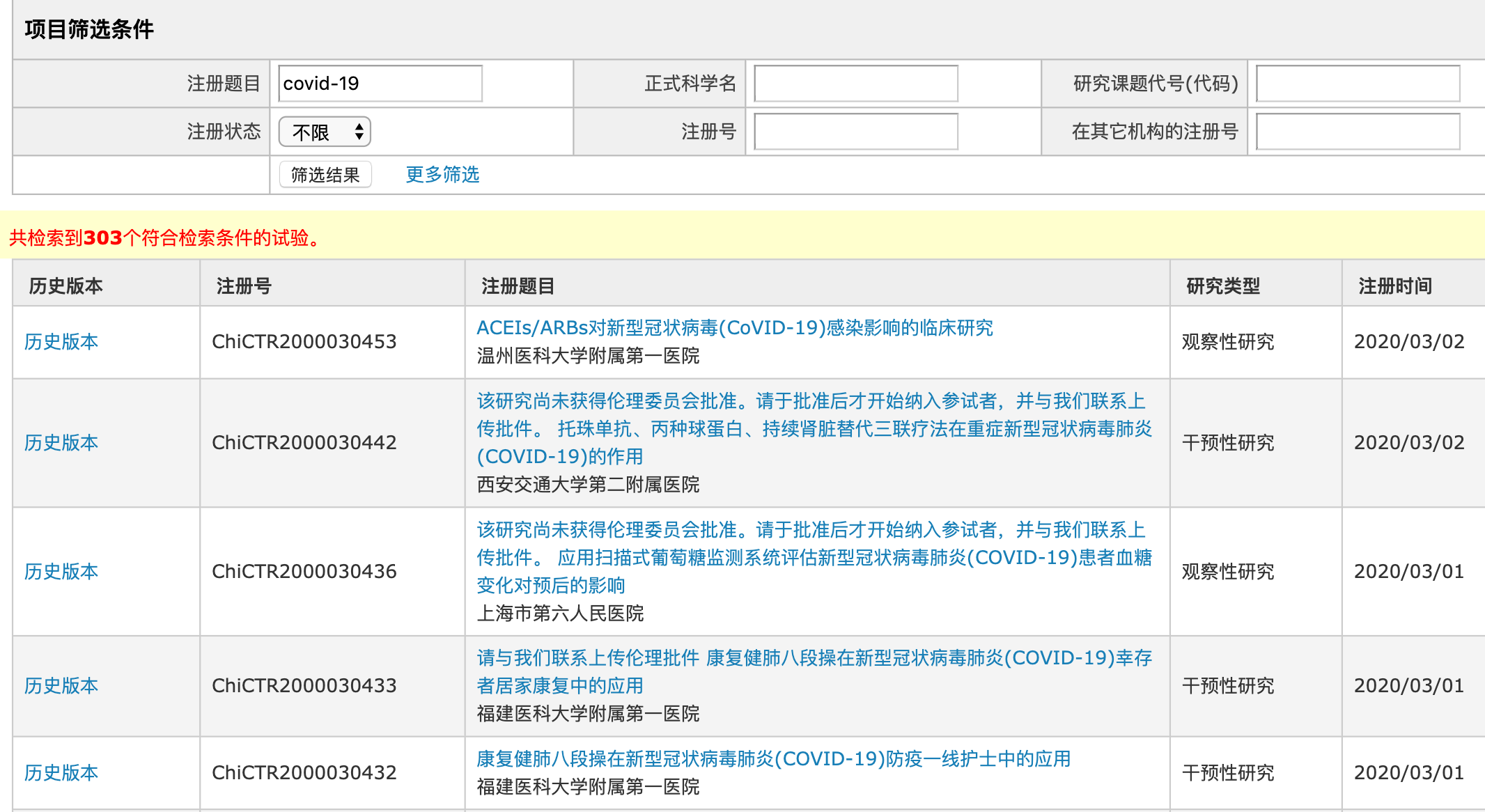
如武汉大学中南医院开展的“大豆水提物对新型冠状病毒肺炎 (COVID-19) 的临床治疗研究”被戏称为“豆浆疗法”;华中科技大学同济医学院附属同济医院主持的400例确诊病人参与的 “双黄连口服液治疗新型冠状病毒肺炎(COVID-19)有效性和安全性的随机、开放、平行对照、多中心临床试验 ”;武汉人民医院一项名为“雾化吸入喜炎平注射液治疗新型冠状病毒肺炎 (COVID-19)的临床研究”;甚至一些太极拳、六字诀的临床试验。
综合各类媒体报道与文献资料不难发现,这些中成药、中药注射液、中医、豆浆其基本都缺乏基本的治疗新冠肺炎或是抑制新冠病毒的概念验证,甚至连体外细胞实验都未曾开展。
当然,缺乏概念验证也并非“中字头”药物和疗法所独有,许多抗生素、干细胞等药物/疗法也同样如此。
一个典型案例便是武汉金银潭医院所注册开展的一项“探究PD-1单抗(卡瑞利珠单抗)和胸腺肽用于伴随淋巴细胞减少的2019新型冠状病毒感染重症肺炎患者的疗效”的临床试验。
PD-1单抗是当下最热门的肿瘤药物,但多位业内人士对其是否存在治疗新冠肺炎效果提出质疑。原因在于,从药物机理上看,PD-1单抗的作用主要是激活与恢复T细胞功能,但新冠病毒在感染人体过程中不仅并没有PD-1的参与,并且被激活后的T细胞也存在加重细胞因子风暴的可能,后者被认为是目前许多危重症新冠肺炎患者的重要死亡原因之一。
这也就不奇怪会有行业内人士质疑使用PD-1治疗“思路是不是反了”,“可能反而会加重患者病情”。
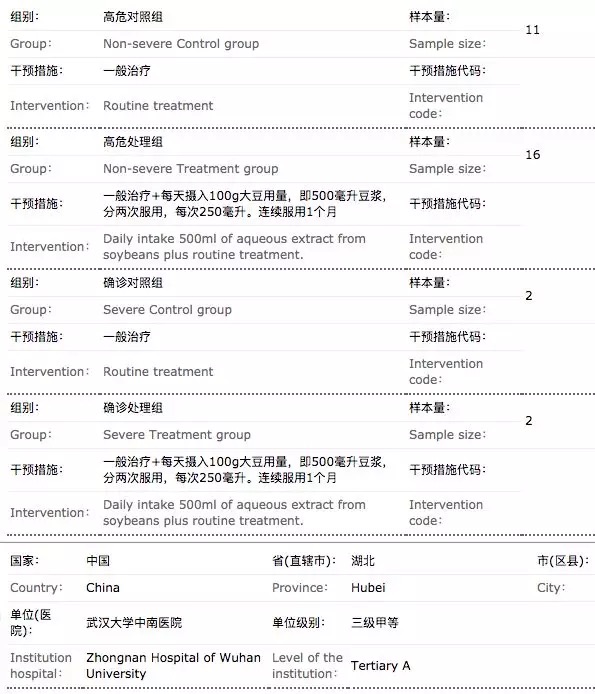
在临床试验方案设计上,也出现了诸多质量不高的案例。最典型的依然是“豆浆疗法”,其不仅整个临床试验各队列入组患者人数都很少,令人匪夷所思的是对照组仅设置为2人,无论是从概率还是统计学角度,都难以看出2人的对照组的效力。而类似的仅设计几十例的临床试验并不在少数。
不过,中国临床试验网站上最新检索发现,这一临床研究申请状态已变更为“研究者撤销”。

“临床中相当一部分的研究样本量很小,研究设计开放性为多,无法提供高质量的临床证据指导用药。”刘熠指出。
她认为,新冠肺炎的临床试验设计具备相当的挑战性:首先从病理生理到疾病诊断上看,目前对于疾病本身还处在一个逐渐认识的过程,如缔脉生物参与的几个新冠肺炎临床试验方案便是“一边在修改方案,另外一边卫健委就又发布了新版的诊疗方案”;其次,不同严重程度患者的治疗目的不同,需要分别进行讨论,用不同的临床评估指标来判断;最后,针对疫情特殊的临床诊疗状态,在研究设计中也需要充分考虑可行性和可操作性。
“一个好的临床试验方案要写两三百页,涉及如何分组,随机、安全风险控制、试验背景。”柯楠介绍,其所在的团队光设计方案就需要4个专职人员至少工作两三个月。他表示,随机对照试验(RCT)依然是临床试验的金标准,但目前也确实不是所有的研究者都有能力开展,在具备充足理由的情况下,针对新冠肺炎患者也可以开展单臂(即无对照)的、或者是非随机对照的临床试验。
不过,临床试验设计的统计学效力和方法是否准确可行、对于药物剂量、观察指标,临床终点的选择上不仅应可以用统计学方法来比较,还应当对临床具有指导意义,很多时候这些是需要强大的统计师团队来完成的。但是,目前大部分一线医生是否具备这一专业能力,要打上个问号。
此外,在当前疫情的特殊时期,实际操作中还面临着更多困难影响着一项临床试验的质量。曹丽君认为,临床试验有大量基础和常规工作需要完成,而医院往往没有足够的专职人员;很多研究者还有大量的临床一线工作,其精力面对大量的项目管理工作也有限。
实际上,在制药企业开展的临床试验中,往往会聘请临床合同研究组织(CRO)来进行临床试验的设计与运营,但据悉有临床CRO参与的研究者发起的临床试验并不多,这主要是和研究规模、经费和研究者对于临床试验质量的观念有关。
规范还应更详细
毫无疑问,低质量的临床试验方案会产生低质量的研究结果,界面新闻记者此前从业内获悉一项数据:目前国内研究者已对外递交超过150篇待发表研究,但退审的文稿高达90多篇,而美国研究者递交了90多篇文章,却只有十几篇被退稿。而美国目前的可供研究的患者人数也是远低于国内。
而综合各方信息可以大致总结出,行政干预、经费驱动、发表研究的需求以及人情关系等是各类低质量的新冠肺炎临床试验不断被立项、批准、运行的几个主要因素。
更为现实的是,混乱的临床试验也会给疫情之中的患者造成伤害,一些患者如果入组一些明显不合理的药物临床试验,是否反而会耽误治疗、研究者又能否做好受试者保护,都是未知数。
这些乱象并非没有引起重视。
世界卫生组织(WHO)此前召开的全球研究创新论坛上就曾对中国新冠病毒相关临床试验提出意见。WHO首席科学家Soumya Swaminathan表示,如果中国的试验设计没有严格的研究参数标准,例如对照组、随机分组和临床结果的衡量标准,那么这些努力将是徒劳的。
而最新发布的《关于规范医疗机构开展新型冠状病毒肺炎药物治疗临床研究的通知》也在事实上对研究者发起的新冠肺炎药物临床试验提出了规范要求,包括限制“所使用的药品应为已上市药品”、要求给药剂量“不超过现有药品说明书的用法用量,预期人体内药物浓度可以达到体外实验换算到人体有效浓度”、明确医疗机构是临床研究的责任主体、提出“医院根据需要可聘请独立于药品供应方、参与临床研究工作的医务人员和患者的IDMC(独立数据监测委员会),在临床研究结束之前定期对研究进展情况进行评判”等。
不过,对于详细的药物筛选、临床终点等具体问题,上述通知并未作出规范指导。
刘熠建议,应当集中资源集中解决重大的临床诊疗问题,可以由监管机构、临床专家和公共卫生专家联合推荐高优先级选题;参照埃博拉疫情期间的国际操作,尽快建立研究多种疗法、多种疾病或两者的母方案(master protocol), 推动标准化的临床研究方案,从而摒弃一些在研究设计上就有缺陷的临床试验。
她还表示,对研究者和伦理委员会的素质和资质需要有严格的审查和管控。在对全部研究者发起的临床试验无法做到统一监管的条件下,可以制定高风险临床试验的监管办法。对临床研究的立项、实施、数据管理、结果公布等,以及配套的硬件软件给出明确指引。最后,对临床试验违规或造假等造成不良后果的处罚应有法律支持。
评论